





| World Journal of Nephrology and Urology, ISSN 1927-1239 print, 1927-1247 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Nephrol Urol and Elmer Press Inc |
| Journal website http://www.wjnu.org |
Case Report
Volume 5, Number 3, September 2016, pages 67-70
A Rare Cause of Polyuria and Polydipsia in a Patient With Cystic Renal Disease: Maturity-Onset Diabetes of the Young Type 5
Hulya Nalcacioglua, c, Belma Haliloglub
aPediatric Nephrology Department, Diyarbakir Childrens’ Hospital, Diyarbakir, Turkey
bPediatric Endocrinology Department, Diyarbakir Childrens’ Hospital, Diyarbakir, Turkey
cCorresponding Author: Hulya Nalcacioglu, Department of Pediatric Nephrology, Kayseri Educational and Research Hospital, 38060 Kayseri, Turkey
Manuscript accepted for publication July 15, 2016
Short title: Polyuria and Polydipsia
doi: http://dx.doi.org/10.14740/wjnu272w
| Abstract | ▴Top |
Hepatocyte nuclear factor-1β (HNF-1β) is a transcription factor that is responsible for the development of kidney, pancreas, liver and genitourinary tract. Affected individuals may present a variety of renal developmental abnormalities and/or maturity-onset diabetes of the young type 5 (MODY 5). Here we report a boy with autosomal recessive polycystic kidney disease (ARPKD) diagnosed in neonatal period who developed insulin-dependent diabetes at the age of 11. He presented with poliuria and polydipsia. The diagnosis of ARPKD was made in neonatal period based on the findings of large hyperechogenic kidneys in antenatal ultrasound and no history of renal disease in parents. Laboratory investigations revealed hyperglycemia, glycosuria, and a reduced glomerular filtration rate (GFR). Based on autoantibody-negative diabetes and low-dose insulin requirement in addition to renal anomalies, he was suspected to have MODY 5. Genetic studies identified a known heterozygous HNF1B gene mutation (S148L) compatible with an MODY 5 phenotype. As a result, MODY 5 should be considered in children with developmental kidney disease and hyperglycemia. Also HNF-1β mutations should be suspected in patients with undefined cystic kidney disease especially when associated with other systemic findings as in our case.
Keywords: Cystic kidney disease; Maturity-onset diabetes of the young; Hepatocyte nuclear factor-1β
| Introduction | ▴Top |
Renal cystic disease may be determined during fetal life or in childhood primarily with ultrasonography [1, 2]. In cases of moderately enlarged hyperechoic kidneys (+2 SD for age) during the second and third trimester, diagnoses of autosomal recessive polycystic kidney disease (ARPKD), autosomal-dominant polycystic kidney disease (ADPKD) and transcription factor 2 (TCF-2) gene mutation related nephropathy should be considered [2, 3]. TCF-2 mutations may be involved in a wide spectrum of renal phenotypes that include renal cysts/renal hyperechogenicity, renal hypoplasia/cystic dysplasia, dysmorphic calyces, urinary tract dilatation, vesico-ureteric reflux, horseshoe kidney and renal agenesis [4-7].
Hepatocyte nuclear factor-1beta (HNF-1β) encoded by the TCF2 gene is involved in the embryonic development of liver, kidney, intestine and pancreatic islets and plays a role in the specific regulation of gene expression of these organs [4, 5]. Also, HNF-1β-regulated genes cause extra-renal abnormalities such as genital malformations (uterus agenesis, bicornuate uterus, agenesis of deferens channels, and hypospadias), abnormal liver function, hypomagnesaemia, hyperuricaemia and early-onset gout [4-6].
Renal cysts are the most commonly observed clinical feature in HNF-1β-associated disease and heterozygous mutations in HNF-1β are additionally known to be responsible for the early-onset diabetes mellitus. This combination led to the description of renal cysts and diabetes (RCAD) syndrome, also referred to as maturity-onset diabetes of the young type 5 (MODY) [7-9]. Here we report a patient with ARPKD diagnosed in neonatal period who developped insulin-dependent diabetes mellitus at the age of 11.
| Case Report | ▴Top |
An 11-year-old boy who has been followed up with a diagnosis of ARPKD at another hospital since 3 months of age applied to the nephrology department with a complaint of polyuria and polydipsia over 3 months. His parents also noted that he had fatigue and weight loss in the same period. He was the third child of consanguineous and healthy parents. His birth weight was 2,200 g after 38 weeks of gestation. At 22 weeks of gestation, fetal ultrasound revealed normal findings other than large hyperechogenic kidneys. He received no specialist follow-up in the first few postnatal months.
On referral to a local nephrologist at 3 months of age, he was found to have elevated serum creatinine levels (1.9 mg/dL) with metabolic acidosis. Ultrasonography showed slightly enlarged kidneys (longitudinal diameter: right 63 mm and left 61 mm), loss of corticomedullary differentiation, and diffuse hyperechogenicity with bilateral multiple small (2 - 6 mm) renal cysts of predominantly cortical distribution. The liver showed parenchymal heterogeneity. He was hospitalized and conservative therapy with nutritional management and oral bicarbonate supplementation were provided according to biochemical results. Liver function tests were all normal. Careful evaluation did not identify any extra-renal malformation. There was no family history of renal disease but he had two type 2 diabetic relatives (maternal uncle and paternal aunt). Ultrasound of the father (at the age of 38 years) and the mother (at the age of 37 years) showed normal kidneys and liver. Thus, the diagnosis of ARPKD was considered based on the prenatal renal ultrasound findings and negative family history. He was discharged from the hospital and referred to pediatric nephrology follow-up. The further clinical follow-up was irregular. At 8 years of age, renal ultrasonography showed hyperechogenic kidneys smaller than expected for age (right 59 mm and left 60 mm) with bilateral small cortical cysts (≤ 5 mm).
At 11 years of age, he was admitted to our nephrology clinic due to polyuria and polydipsia. On admission, he was dehydrated and seemed tired and weak. His height was 130 cm (< 3th percentile) and weight was 24.3 kg (< 3th percentile). Vital signs were normal. Urinalysis was significant for 3+ glucosuria and 1+ proteinuria. Serum biochemistry revealed glucose 391 mg/dL, urea 87 mg/dL, creatinine 1.95 mg/dL, Na 132 mmol/L, K 5.5 mmol/L, Ca 9.7 mg/dL, P 5.2 mg/dL, albumin 4. 6 g/dL and metabolic acidosis. Abdominal ultrasound showed bilateral small kidneys, increased parenchymal echogenicity and cysts and no evidence of portal hypertension or splenomegaly-hepatomegaly. No evidence for hyperglycemia was found to date in the patient. HbA1c level was 9.1% (N: 4.3-5.6%) and also islet cell antibodies (ICAs) and glutamic acid decarboxylase (GAD) antibodies were negative. Diabetes mellitus was diagnosed and insulin therapy was commenced. His metabolic control was very well with low-dose insulin treatment (0. 3 U/kg/day). Based on autoantibody-negative diabetes, low-dose insulin requirement in addition to renal anomalies, he was suspected to have MODY 5. Genetic testing revealed a known missense heterozygous mutation (p.S148L, c.443 C>T) in the HNF-1β gene. This result confirmed the diagnosis of HNF-1β MODY. The other extra-renal findings of HNF-1β were checked. The liver enzyme levels, magnessium level and fecal elastase were normal. Also, physical examination and radiologic images did not detect any genital tract abnormalities, pancreatic hypoplasia or parathyroid adenoma/hyperplasia. During the follow-up period of 6 months, his renal function stayed stable at stage 3 chronic kidney disease (glomerular filtration rate (GFR), 47 mL/min/1.73 m2) and his insulin requirement and HbA1c level were 0.5 U/kg/day and 6.9%, respectively.
| Discussion | ▴Top |
In the present paper, we reported a patient with ARPKD who presented with autoantibody-negative diabetes at the age of 11 years caused by a heterozygous HNF-1β mutation. MODY 5 paved the way to the accurate diagnosis.
Clinical presentation of renal involvement in utero was highly suggestive of severe polycystic kidney disease. Given increased kidney size, multiple cysts with significant renal failure and the absence of family history suggested a diagnosis of ARPKD [2, 10, 11]. However, the size of kidneys which are not enlarged relative to body size, the lack of hepatic fibrosis and portal hypertension during the follow-up mainly by ultrasound in our patient was puzzling. In ARPKD macrocysts are not routinely present at birth and kidney size stabilizes or may decrease over time and these patients do not show progressive macrocystic enlargement as in ADPKD. In addition, some patients may lack specific symptoms and results of imaging studies and liver tests may be in the normal range [10-12]. Although diabetes was diagnosed in our patient at second decade, further findings of endogenous insulin secretion (low-dose insulin requirement) and the absence of autoantibodies for diabetes and cystic kidneys prompted consideration of the diagnosis of MODY [8]. The association of polycystic kidney disease and MODY phenotype best fit RCAD, also referred to as MODY type 5 prompted genetic testing of HNF-1β [8, 9].
MODY has been described in 58% of reported HNF-1β mutation carriers, with mean age of diagnosis of 26 years [7-9]. In this context, renal involvement seems to be the first manifestation of MODY 5 and some of these features can be identified by antenatal ultrasound [5, 6, 13]. The most frequent antenatal presentation is bilateral hyperechogenic kidneys with normal or increased size [6, 14]. In our patient, bilateral enlarged hyperechogenic kidneys as the common assumption of ARPKD were already confirmed by antenatal US and the presence of associated extrarenal abnormalities as MODY 5 led us to consider the diagnostic accuracy and study direct genetic testing. Remarkably, the phenotype of bilateral hyperechogenic kidneys incidentally found during pregnancy in our patient with renal failure can be explained by HNF-1β mutation. Decramer et al [14] screened on the diagnosis of fetal bilateral hyperechogenic kidneys and found HNF-1β mutations in 29% of the 56 newborn infants. HNF-1β appears to be a main regulator in the expression of genes of which mutations are responsible for cystic kidney diseases (Nphp1, polaris, Umod, Pkhd1, and Pkd2) [5]. The finding of polycystic kidneys is not unpredicted in patients with TCF2 mutation. Previous studies have shown that HNF-1β directly regulates Pkhd1 gene expression and established a novel link between two renal cystic diseases MODY5 and ARPKD [13, 15].
Mutations in HNF-1β are inherited in an autosomal dominant pattern, although de novo mutations do occur for a third to half of all cases explaining lack of family history in many patients in the reports [4, 5, 7]. Mutations may involve heterozygous deletion of the whole gene or small mutations including missense, non-sense, frameshift and splice site mutations have been found in different domains [5, 7]. Several studies have not found a clear genotype phenotype correlation in terms of the severity and/or type of renal disease [3, 13, 16, 17]. The mutation in our case (p.S148L (c.443C>T)) was heterozygous and located in exon 2 within the pseudo-POU domain of the gene that determines the target sequence specificity of the HNF-1β molecule. The parents had no history or clinical symptoms related to diabetes or kidney disease. We could not perform genetic analysis in any other family member due to fiscal problems. Therefore, suggesting S148L mutation in our patient to represent a de novo mutation is highly speculative. To date, together with our case, six patients with S148 mutations have been reported with highly different phenotypes (Table 1) [3, 13, 16, 17]. It thus seems that this specific region may present a genetic hot spot for mutations in HNF-1β.
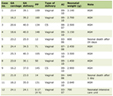 Click to view | Table 1. Case Series of S148 HNF-1β Mutations |
In conclusion, we present a patient with a heterozygous mutation in the HNF-1β gene resulting in a phenotype of severe renal involvement manifested at birth and early-onset diabetes with β-cell dysfunction. MODY 5 should be considered in the differential diagnosis of children with developmental kidney disease and hyperglycemia. Also HNF-1β mutations should be suspected in patients with undefined cystic kidney disease especially when associated with other systemic findings as in our case.
Disclosure
The authors declare that they have no potential conflicts of interest and financial disclosure.
| References | ▴Top |
- de Bruyn R, Gordon I. Imaging in cystic renal disease. Arch Dis Child. 2000;83(5):401-407.
doi pubmed - Avni FE, Hall M. Renal cystic diseases in children: new concepts. Pediatr Radiol. 2010;40(6):939-946.
doi pubmed - Edghill EL, Bingham C, Ellard S, Hattersley AT. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet. 2006;43(1):84-90.
doi pubmed - Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat Rev Nephrol. 2015;11(2):102-112.
doi pubmed - Verhave JC, Bech AP, Wetzels JF, Nijenhuis T. Hepatocyte Nuclear Factor 1beta-Associated Kidney Disease: More than Renal Cysts and Diabetes. J Am Soc Nephrol. 2016;27(2):345-353.
doi pubmed - Ulinski T, Lescure S, Beaufils S, Guigonis V, Decramer S, Morin D, Clauin S, et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol. 2006;17(2):497-503.
doi pubmed - Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, Lindner T, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17(4):384-385.
doi pubmed - Timsit J, Bellanne-Chantelot C, Dubois-Laforgue D, Velho G. Diagnosis and management of maturity-onset diabetes of the young. Treat Endocrinol. 2005;4(1):9-18.
doi pubmed - Bingham C, Hattersley AT. Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol Dial Transplant. 2004;19(11):2703-2708.
doi pubmed - Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics. 2014;134(3):e833-845.
doi pubmed - Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, Consugar MB, Milliner DM, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006;85(1):1-21.
doi pubmed - Gresh L, Fischer E, Reimann A, Tanguy M, Garbay S, Shao X, Hiesberger T, et al. A transcriptional network in polycystic kidney disease. EMBO J. 2004;23(7):1657-1668.
doi pubmed - Yorifuji T, Kurokawa K, Mamada M, Imai T, Kawai M, Nishi Y, Shishido S, et al. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: Phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1beta gene due to germline mosaicism. J Clin Endocrinol Metab. 2004;89(6):2905-2908.
doi pubmed - Decramer S, Parant O, Beaufils S, Clauin S, Guillou C, Kessler S, Aziza J, et al. Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J Am Soc Nephrol. 2007;18(3):923-933.
doi pubmed - Hiesberger T, Bai Y, Shao X, McNally BT, Sinclair AM, Tian X, Somlo S, et al. Mutation of hepatocyte nuclear factor-1beta inhibits Pkhd1 gene expression and produces renal cysts in mice. J Clin Invest. 2004;113(6):814-825.
doi pubmed - Mayer C, Bottcher Y, Kovacs P, Halbritter J, Stumvoll M. Phenotype of a patient with a de novo mutation in the hepatocyte nuclear factor 1beta/maturity-onset diabetes of the young type 5 gene. Metabolism. 2008;57(3):416-420.
doi pubmed - Edghill EL, Bingham C, Slingerland AS, Minton JA, Noordam C, Ellard S, Hattersley AT. Hepatocyte nuclear factor-1 beta mutations cause neonatal diabetes and intrauterine growth retardation: support for a critical role of HNF-1beta in human pancreatic development. Diabet Med. 2006;23(12):1301-1306.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Nephrology and Urology is published by Elmer Press Inc.