





| World Journal of Nephrology and Urology, ISSN 1927-1239 print, 1927-1247 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Nephrol Urol and Elmer Press Inc |
| Journal website http://www.wjnu.org |
Case Report
Volume 000, Number 000, January 2016, pages 40-47
Gitelman Syndrome: An Important Cause of Severe Hypokalemia and Periodic Paralysis
Friska Sinagaa, Ridho M. Naibahoa, Wilfried Herdin Sibueaa, b
aDepartment of Internal Medicine, Tebet Hospital, Jakarta, Indonesia
bCorresponding Author: Wilfried Herdin Sibuea, Department of Internal Medicine, Tebet Hospital, Letjen M.T. Haryono 8, South Jakarta, 12810, Indonesia
Manuscript accepted for publication May 05, 2016
Short title: Gitelman Syndrome
doi: http://dx.doi.org/10.14740/wjnu270e
| Abstract | ▴Top |
Gitelman syndrome is a relatively rare renal tubular disorder. Though it has been reported as a recessively inherited disorder, sporadic cases have also been reported. Traditionally, Gitelman syndrome is considered as benign or mild tubulopathy. We present the case of an 18-year-old male patient with severe hypokalemia and periodic paralysis. Subsequent laboratory investigation revealed renal wasting hypokalemia, metabolic alkalosis, secondary hyperaldosteronism, hypomagnesemia and hypocalciuria, indicating that the patient might have had a renal tubular disorder. The confirmation of Gitelman syndrome was determined by evaluating tubular function using thiazide and furosemide challenge test. Genetic study was bypassed due to our technical unavaibility. Treament included magnesium aspartate/potassium aspartate, potassium chloride tablets and potassium-sparing diuretic. We are presenting our case seeing that Gitelman syndrome is not a syndrome to be overlooked as it bears a risk of severe complications. Gitelman syndrome may present in adulthood and should be borne in mind in the diagnosis of hypokalemia.
Keywords: Gitelman syndrome; Renal tubular disorder; Hypokalemia; Periodic paralysis
| Introduction | ▴Top |
Gitelman et al [1] in 1966 described a different myriad of patients with renal tubulopathy. Hypokalemia, hypomagnesemia due to renal defective mechanism to conserve potassium and magnesium ions, metabolic alkalosis and hypocalciuria are the hallmark features of the reported cases. All share the similar characteristics with Bartter syndrome, but the hypomagnesemia and normocalcemic-hypocalciuria are the typical that distinguish Gitelman syndrome from the “classical” Bartter syndrome [1-3]. Gitelman syndrome, which is attributed to defects of the transportation system in the distal convoluted tubule (DCT), is recognized linked to the gene encoding the thiazide-sensitive Na-Cl cotransporter (NCCT) located on chromosome 16q. Bartter syndrome, on the other hand, is caused by mutations in the transporters in the thick ascending of loop of Henle [4]. Sodium chloride wasting and subsequent activation of renin-angiotensin-aldosterone system (RAAS) explain the clinical phenotype of this rare tubulopathy [2, 3]. The definitive diagnosis of Gitelman syndrome lies in the documentation of a homozygous/compound heterozygous mutation in the NCCT gene by direct sequencing, which is not always available in every institution [5].
The clinical phenotype of Gitelman syndrome patient is highly heterogenous in terms of age of presentation, severity of biochemical abnormalities and clinical manifestation [6]. Recently, we diagnosed a patient as Gitelman syndrome by evaluating laboratory results and distal nephron function test. So that the traditional view to NCCT mutation results in only mild salt-wasting, Gitelman syndrome is considered a mild variant of salt-losing tubulopathy [2-4, 7]. However, our case below presents with severe hypokalemia and paralysis. At first glance, he was misdiagnosed as hypokalemic periodic paralysis and Gitelman syndrome has not been considered. The diagnosis was established after an effort on laboratory examination to find the cause by doing comprehensive workup.
| Case Report | ▴Top |
An 18-year-old Indonesian male who had experienced 3 years episodes of generalized weakness along with diffuse muscle aches and cramp was admitted to our hospital in January 2009. The usual weakness was precipitated by resting after physical exertion and came into a stereotypical fashion: it began with numbness in proximal part of extremities, tiredness, fatigue and then progressively worsened until the patient was unable to walk. Each episode usually lasted a few hours to couple of days with residing weakness. Between attacks, his only symptoms were nocturia and occasionally joint pain. One year earlier, he was found to be hypokalemic when he visited a private clinic for evaluation of similar weakness. Complete data were unavailable, except that the last consultation on December 2008 disclosed a potassium concentration of 3.0 mEq/L and that he received potassium supplementation. However, hypokalemia was never completely corrected and he was referred for diagnostic stay.
The patient was non-smoker, and consumed no alcohol or any herbal products. There was no vomiting or diarrhea, and he had no past history for any medical prescription including diuretics or recreational drugs. His parents noted that he had a habit of salty foods such as pickles, salted fish, and potato crips. Perinatal history was unremarkable and experienced a normal delivery with birth weight of 3,100 g. He grew up normally with no childhood history of tetany or similar symptoms. No other family member had a similar illness and there was no history of parental close-family marriages.
On admission, he was alert, look very weak and lying listlessly. Physical examination showed a thin man (he was 1.64 cm in tall and weighed 48 kg) with normal in-patient blood pressure. Throughout his course in the hospital, his blood pressure measurements were around 100 - 110 over 65 - 70 mm Hg. There was no eye or other signs suggestive of thyrotoxicosis and his thyroid gland was not enlarged. Cardiac examination showed a regular rate of 70 beats/min in the supine position. Chest expansion was limited. Examination of the abdomen revealed no tenderness or masses but diminished bowel sounds. There was muscular atrophy and both legs fell laterally (Fig. 1). Manual motoric muscle test revealed 1/5 weakness in his neck and extremities with diminished deep tendon reflex in upper and lower extremities. All nervi cranialis remain intact. There were no sensory deficits and pathological reflexes were absent bilaterally. Trousseau’s and Chvostek’s sign was negative. He was clinically euvolemic as skin turgors were found to be normal. The rest of the general physical examination including spine was also normal.
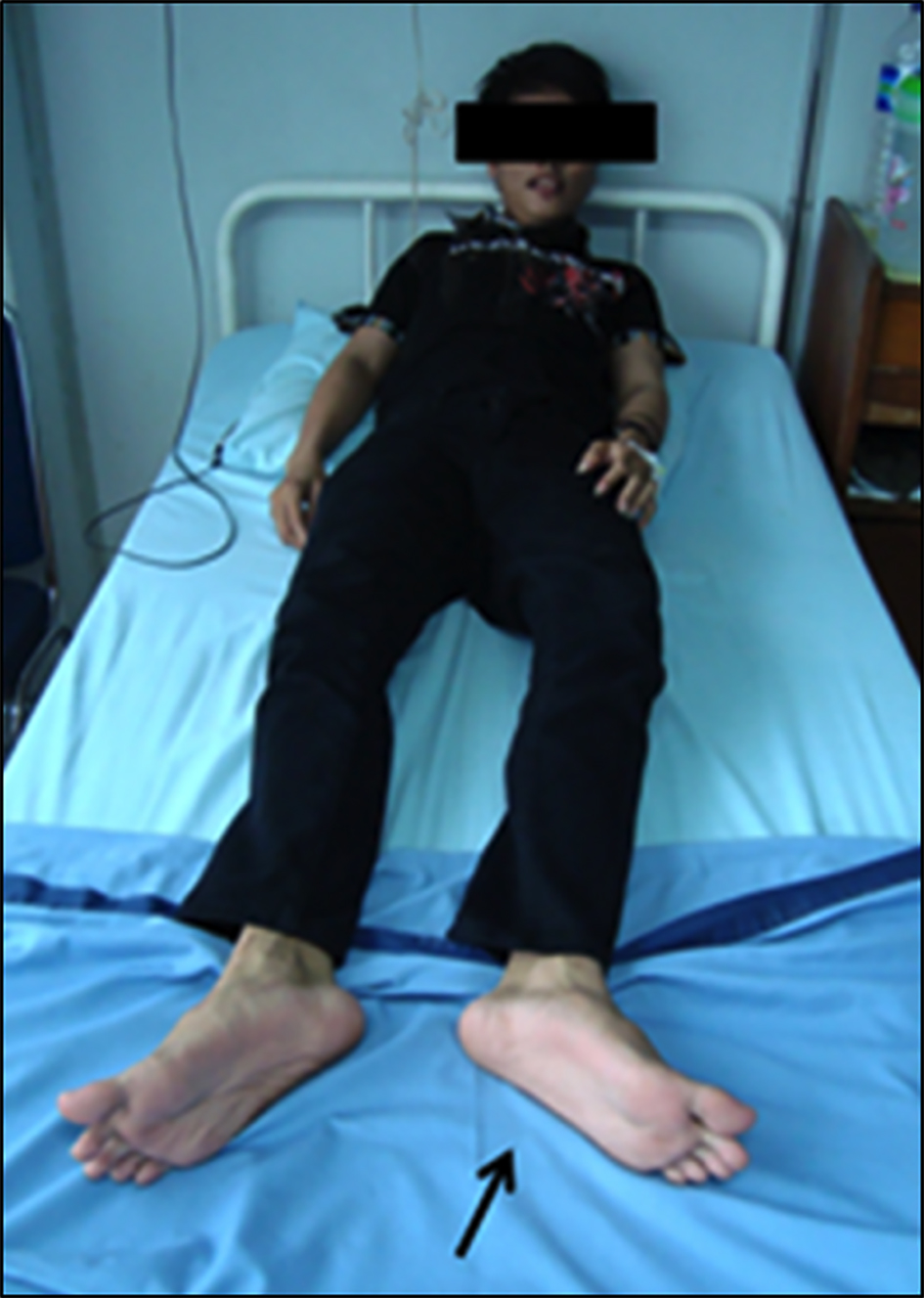 Click for large image | Figure 1. The photograph of the patient, please notice on foot weakness and both legs fell laterally (black arrow). |
The results of laboratory workup are shown in Table 1 with outstanding findings of severe hypokalemia (K = 1.5 mEq/L), metabolic alkalosis (pH = 7.55; HCO3 = 31.5 mMol/L; PCO2 = 32.2 mm Hg), hypomagnesemia (Mg = 1.3 mg/dL) with hypocalciuria (urinary calcium excretion ranged between 19.2 and 16.9; N = 50 - 400 mg/g creatinine). Creatine phosphokinase (CPK) level was 1,236 IU/L (maximum at day 2 with CPK level 1,977 IU/L). Urinalysis showed a pH of 7.0 and specific gravity of 1.015. No glucosuria and hemoglobinuria were detected but trace of proteinuria was present. Glomerular filtration rate (GFR) expressed as creatinine clearance was normal. The urinary electrolyte values revealed sodium excretion was 402 mEq/L, chloride was 281 mEq/L and potassium was 41.8 mEq/L per 24 h. The urinary Ca/creatinine ratio was low at 0.04 mg/mg (N ≥ 0.2) [8]. The fraction excretion of potassium (FEK) was 29.4% (N = 4-16%, mean value 8%) [9] and high transtubular potassium gradient (TTKG) suggested that renal potassium wasting was responsible for the hypokalemia [10]. This findings indicated that the patient might have had renal tubulopathy and prompted us to examine distal tubular function.
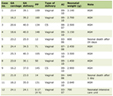 Click to view | Table 1. Biochemical Investigation in the Hospital |
To identify the impaired portion of the renal tubules, we performed renal clearance studies using two diuretics, that is, thiazide and furosemide. These studies were performed using the protocol described earlier by Kageyama et al [11] and informed consent was obtained from the patient. For thiazide-loading test, 50 mg (about 1 mg/kg of body weight) of hydrochlorothiazide was orally administered; for the furosemide loading test, 40 mg of furosemide was given intravenously to the patient. Furosemide and thiazide tests were carried out in two phases with 7 days apart. Changes in fractional excretion of solutes (for sodium, potassium and chloride), free-water clearance (CH2O), chloride clearance (CCl), distal fractional chloride reabsorption (DFCR) and fractional distal delivery of chloride (FDDC) were measured. As shown in Table 2, furosemide injection resulted in marked increased in FENa, FEK, and FECl with reduced CH2O, while thiazide did not significantly alter the CH2O but did result in increased fractional excretion of solutes but to a lesser extent than caused by furosemide. DFCR was markedly decreased by furosemide administration (from 83.4% to 7.2%), whereas thiazide had only a minor effect on this parameter.
Click to view | Table 2. Data on Renal Clearance Studies |
The subsequent data on the electrocardiogram (Fig. 2) showed a normal sinus rhythm with QRS rate of 80 - 90 beats/min, QRS complex duration of 0.1 s, ST-segment depression in leads V1 through V5 with flattened T, prominent U waves and prolonged corrected QT interval (QTc = 580 ms). Chest X-ray was normal. Features of his electromyography and nerve conduction study performed after resolution of hypokalemia were similar to those observed in periodic paralysis. Ultrasound of the kidney and suprarenal was unremarkable. There was no nephrocalcinosis. Magnetic resonance imaging of the whole abdomen demonstrated neither adrenal hyperplasia nor intraabdominal mass. Endocrine examination disclosed high plasma renin activity (PRA) and plasma aldosterone concentration (PAC). The PAC-to-PRA ratio was 5.22. Other endocrine investigations related to hypokalemic paralysis included thyroid function test such as triiodothyronine (T3), thyroxine (T4) and thyroid stimulating hormone (TSHs) which were within the reference range.
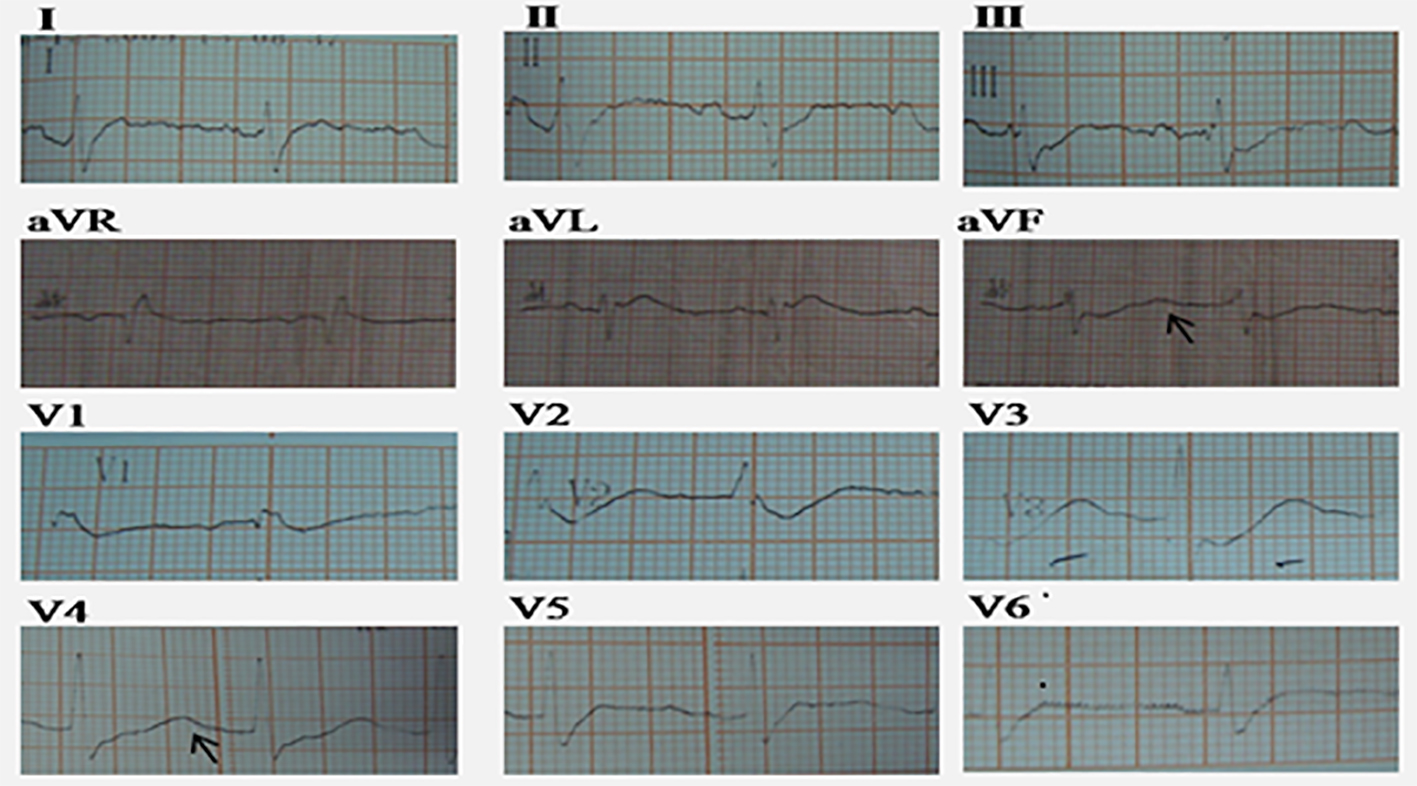 Click for large image | Figure 2. Electrocardiograph of the patient note the typical hypokalemic pattern of myocardial repolarization disturbance with depressed ST segment, wide and bizzare T wave deflection, the occurrence of U wave (black arrow) and prolonged QTc interval (580 ms). |
He was diagnosed as Gitelman syndrome based on anamnestic data, clinical picture, repeated laboratory documentation of hypokalemia, hypomagnesaemia, metabolic alkalosis, hypocalciuria, and secondary hyperreninism hyperaldosteronism. The diagnosis was supported on the basis of DCT defect evidenced by blunted thiazide response. In this patient, the thick ascending limb of Henle was intact as demonstrated by appropriate natriuretic response after loop diuretic injection [4].
The patient was initially treated with vigorous volume replacement with normal saline (3 - 4 L/day), intravenous potassium chloride (reaching 120 mEq/day) and placed also on an oral magnesium replacement due to observed hypokalemia and hypomagnesemia. On the second hospital day, the patient began to have significant movement and progressively improved muscle strength with concomitant increased in his potassium level. After 1 week, the muscle weakness recovered following a rise in serum potassium, and his CPK had decreased to 314 IU/L which normalized after 3 weeks. Doses of intravenous and oral supplementation of potassium and magnesium were adjusted according to symptoms and serum electrolytes; spironolactone was also added with titration dose started from 50 mg bid. The weakness gradually progressed and manual motoric test improved to 4-5/5 in all extremites. The patient discharge with formulation consisted of 600 mg oral potassium chloride tid, 100 mg spironolactone bid and 300 mg magnesium aspartate/potassium aspartate tid (maximum serum potassium and magnesium were 3.6 and 2.1 mEq/L, respectively). He was also counselled regarding the importance of regular follow-up and need for being complaint with the treatment and exclusively scheduled for six monthly visits course in June 2009. Serum electrolytes of the patient’s parents and his older brother were normal.
| Discussion | ▴Top |
Hypokalemia is a common clinical problem in endocrinologists’ and nephrologists’ or general internists’ practice. The causes of hypokalemia in most cases were obvious from the patient’s history and in the majority of cases can be traced by a simple investigation. Nevertheless, its etiology in some cases is not staightforward and demands a workup with sometimes surprising conclusions [12]. Gitelman syndrome is also known as familial hypokalemic hypomagnesemia, because hypokalemia is the most common presentation [1, 2]. Due to its low prevalence, Gitelman syndrome is rarely considered in the initial differential of weakness and paralysis. The problem of this rare inherited tubulopathy is that overlooked hypokalemia could cause paralysis or even death due to ventricular arrhythmias and cardiac arrest [3]. We will discuss how we reach the diagnosis of Gitelman syndrome and the most likely explanation for its management in our patient.
Four potential mechanisms were thought to responsible for hypokalemia. These include inadequate potassium intake (i.e. anorexia nervosa, long-term hunger), potassium shift from extracellular to intracellular fluid compartment, non-renal loss (i.e. sweating, vomiting and diarrhea), and excess renal potassium losses [11, 13]. Hypokalemia in our patient seemed not to be related to inadequate intake as anorexia nervosa and long-term hunger were not detected. Potassium cellular shift might be related to hypokalemic periodic paralysis, or during treatment with insulin, salbutamol and theophylline, was exclusively excluded by negative history of such medications. Thyrotoxicosis was not the case as thyroid function test was found to be normal. Chronic vomiting was also excluded as the patient had no history of persistent vomit and his urinary chloride was high, consistent with active renal salt loss [11]. The last possibility is therefore increased renal loss that should be confirmed by findings of high urinary potassium excretion.
In our case, hypokalemia was proven to be in renal origin as we can demonstrate high potassium loss (high urinary potassium excretion, high FEK and high TTKG) [13]. The hypothesis of diuretic abuse could also fit well with renal hypokalemia. Urinary diuretic screen was not available in our laboratory; however, this possibility could be promptly excluded by negative history of medication abuse. Nonetheless, the patient kept very low potassium levels while he was in our ward while no outer medication was allowed. Acid-base status is helpful in the rapid approach to a patient with hypokalemic paralysis. Metabolic alkalosis is present in patient with renal potassium wasting syndrome while there is no acid-base alteration in hypokalemic periodic paralysis. Cystic fibrosis was another possibility that was attributed to hypokalemia and alkalosis metabolic; it is typically present in infancy with combination of a failure to thrive and respiratory symptoms [12, 13]. High urinary chloride confirmed renal cause of hypokalemia which makes the possibility of cystic fibrosis very small.
The following features were present in our patient: an age of 18 years, normal blood pressure, hypokalemia, metabolic alkalosis, hypomagnesemia and hypocalciuria. In a patient with known hypokalemic metabolic alkalosis associated with high renin and aldosterone levels and low-normal blood pressure level, the main differential diagnosis could be limited to salt losing-tubulopathies, namely, Gitelman and Bartter syndromes [2-5, 12]. The serum magnesium concentration distinguishes between the two syndromes, being low in Gitelman syndrome and normal in Bartter syndrome [2-4]. Therefore, the most likely hypothesis seemed to be Gitelman syndrome for a number of reasons: presentation in early adulthood, normal growth and hypomagnesemia. The occurrence of hypocalciuria was thought to be pathognomonic of Gitelman syndrome and was used as the distinguished feature between both syndromes [2, 3, 8]. A genetical study would confirm the diagnosis and clarify about clinical and biochemical features of Gitelman syndrome in this case but we could not do gene testing due to lack of facilities [3, 5]. Though this limitation, we do not have many doubts about the diagnosis as we perfomed a renal clearance study to evaluate the tubulopathy.
Theoretically, Gitelman syndrome is likely to represent a defect of the DCT, primary site of action of thiazide. This fact could be used for diagnostic purposes that if patients with Gitelman syndrome take a test dose of thiazide, the normal effect of increased Na, Cl and K diuresis fails to appear. Meanwhile, furosemide acts in the loop of Henle that is anatomically proximal to the DCT will increase Na and Cl diuresis [4]. In our patient, natriuresis and chloruresis responses were blunted following thiazide-loading test as compared to increased values obtained by furosemide. Upon furosemide injection, there was net increase in CCl; due to eventually defect in the DCT, chloride load fails to get reabsorbed. On the other hand, thiazide only mildly increased the FDDC and mildly decreased the DFCR. The moderate degree of NaCl and potassium wasting, the exaggerated natriuresis and kaliuresis after furosemide and blunted thiazide response, all suggest the presence of a primary defect in the DCT, in accordance with the findings of Gitelman syndrome [4, 11, 14]. The reported thiazide test sensitivity is 93% and specificity 100% to discriminate Gitelman syndrome [14].
Bettineli et al [8] formulated clinical diagnostic criteria for Gitelman syndrome. They defined the case of Gitelman syndrome as sets of abnormalities comprised of hypokalemia of renal origin (< 3.6 mmol/L), hypomagnesemia (< 1.58 mg/dL) and hypocalciuria (molar ratio of urinary calcium to creatinine < 0.2) in terms of normal urine concentrating ability and normal GFR. Hypomagnesemia and hypocalciuria give a high probability for the diagnosis [15]. Nakhoul et al [3] also added negative screening for diuretics and recurrent vomiting from the history in the absence of arterial hypertension. The present case consistently fulfilled all these criteria and renal clearance test has support the diagnosis of Gitelman syndrome. He was treated accordingly.
Gitelman syndrome is caused by genetic mutations of SCL12A3 located on q3 of chromosome 16 which encodes NCCT in the distal convoluted tubule (DCT) [5]. The NCCT in the DCT of the kidney was responsible for the 5-7% reabsorption of NaCl [2, 5, 11, 15]. The prevalence of Gitelman syndrome is around 25 cases per 1 million inhabitants [2]. Although there is an autosomal recessive mode of inheritance and the syndrome is well described in siblings [5, 15], many cases appear to be sporadic [2]. Japanese investigators [16, 17] published several reports of patients with sporadic Gitelman syndrome with a compound heterozygote of a known missense mutation in the SLC12A3 gene. Our patient also appears to be a sporadic case as we could not find a similar history in a nearby family member and hypokalemic screening has negative results.
The complete linkage between clinical picture in Gitelman syndrome and SCL12A3 mutations is now recognized that the underlying potassium wasting is mediated by chronic renal salt-wasting and stimulation of RAAS as the consequence of relative hypovolemia [2-5]. At the end of RAAS cascade, increased aldosterone drive in the collecting tubule (Fig. 3) will cause increased sodium reabsorbtion but caused enhanced potassium and hydrogen ions secretion accounting for an attendant hypokalemia and alkalosis seen in affected patients [2]. Symptoms of Gitelman syndrome patients range from asymptomatic, to mild symptoms of cramps and weakness to severe manifestation such as tetany, paralysis and rhabdomyolisis. Salt craving, polydipsia, polyuria, and nocturia are also prominent symptoms [3, 7].
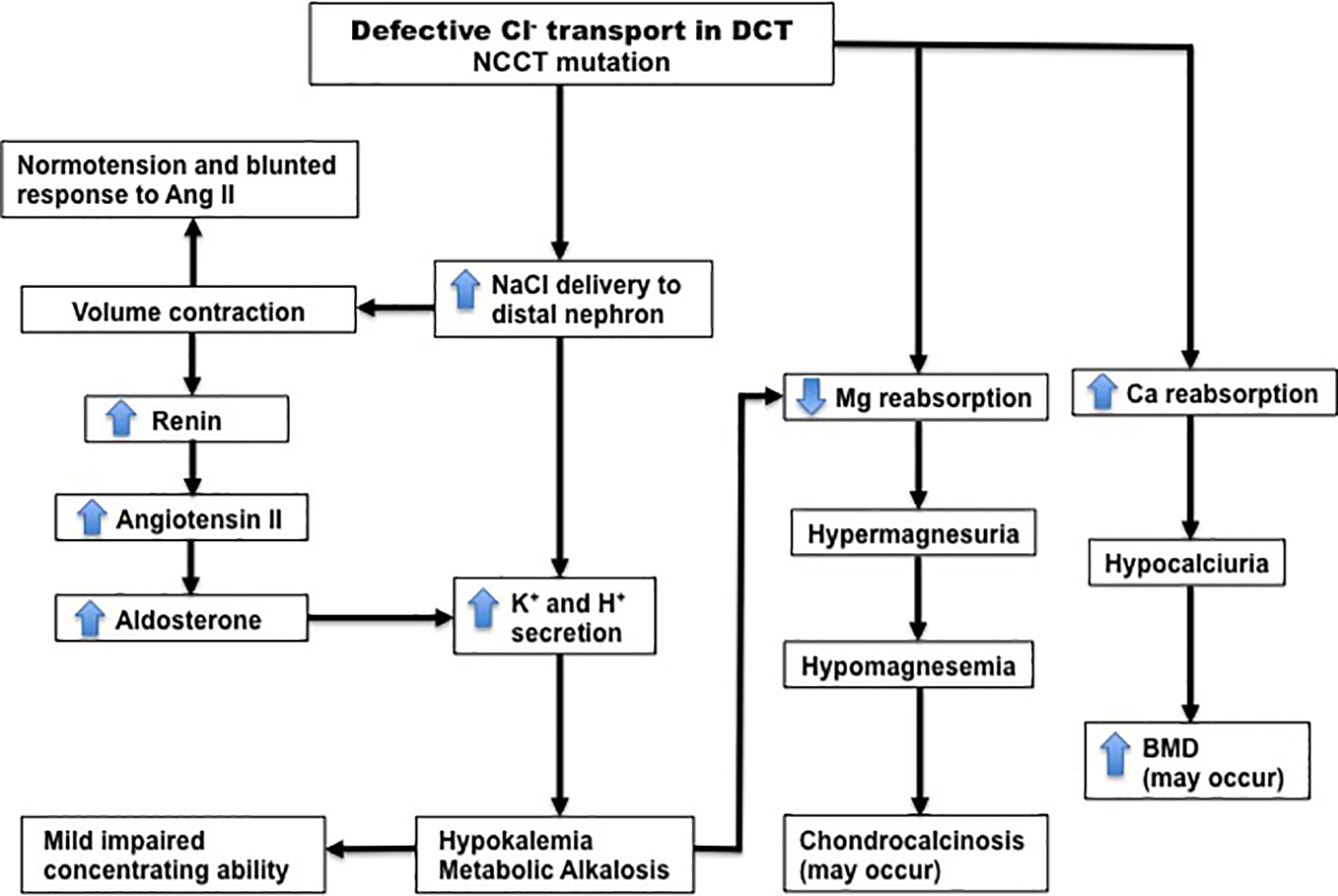 Click for large image | Figure 3. Pathophysiology of Gitelman syndrome. The primary abnormality is defective Cl- reabsorption in the DCT due to inactivating mutations in the NCCT gene. This leads to the main features of the syndrome, which are hypokalemic metabolic alkalosis, secondary hyperaldosteronism, normotension, hypomagnesemia, hypocalciuria and blunted response to angiotensin II. DCT, distal convoluted tubule; NCCT, thiazide-sensitive Na-Cl cotransporter. Source: Shaer AJ. Am J Med Sci 2001, with permission. |
Epidemiologic studies have demonstrated that there is no ethnic predilection for Gitelman syndrome and both sexes are equally affected [3, 6, 7]. However, sex difference may account for some variability because in some series reported by Lin et al [18] that male patients could be affected more severely. Asian population was also reported to have more severe disease with respect to paralysis symptoms [19, 20]. Balavoine et al [6] have demonstrated two mutations of the SCL12A3 gene have seen more severe symptoms and hypokalemia compared to one or no mutation group. The possible explanation for the high frequency of complications found in our patient could be related either to genetic background or probably to more severe potassium and salt wasting which induces more activation of RAAS and deleterious consequences with metabolic pathways.
Hypokalemia is likely to be universal in Gitelman syndrome and its detection is the main clue leading to diagnosis [2-4]. Although hypokalemia is commonly mild in Gitelman syndrome, there are some reported cases having severe hypokalemia [21]. Cruz et al [7] reported that 6% of patients with Gitelman syndrome present with hypokalemic paralysis. Tang et al [19] have demonstrated the presence of myocyte necrosis in a case of Gitelman syndrome with markedly elevated levels of muscle enzymes. Nishihara et al [22] reported case with severe hypokalemia (1.2 mMol/L) who developed rhabdomyolisis and acute renal failure (ARF). To prevent ARF due to rhabdomyolisis, there is no specific treatment except to maintain fluid and electrolyte balance and preserve tissue perfusion. The risk of developing rhabdomyolisis occurs when potassium level is lower than 2 mMol/L [23]. Although rhabdomyolisis was clearly present in our case, rapid rehydration and correction of hypokalemia prevented the development of renal failure.
Our patient’s ECG on admission showed evidence of prolonged QTc interval (580 ms) which resolved (not shown) after treatment of both potassium and magnesium deficiency. Hypokalemia and hypomagnesemia simultaneously prolonged the duration of the action potential of cardiomyocytes and consequently increased the risk of ventricular arrhythmias [2, 3]. Bettinelli et al [24] found that patients presenting QTc interval > 500 ms are at risk of developing such complication. Prolongation of QTc also potentially predisposed to enhanced toxicity of QT-prolonging drugs such as macrolide, antihistamine, cisapride, etc. [24]. These medication should better be avoided for the patient. He should also be discouraged to participate in sporting activities as this may trigger malignant arrhythmias and left ventricular dysfunction [25].
Finally, as with other hereditary diseases, neither the tubular defects in Bartter nor Gitelman syndrome can be fully corrected [2-5]. Current available management has been designed to antagonizing secondary increased in aldosterone secretion and potassium supplementation [2, 3, 7]. Our patient was started on intravenous potassium and magnesium supplementation and subsequently he was discharged on oral potassium and magnesium along with spironolactone. He should receive these medication in his lifetime to avoid atrophy and muscle weakness. It is sometimes not possible to normalize serum level of the electrolyte completely and it is more beneficial to focus attention on the amelioration of patient’s complaints [2, 7]. We advised him to take sufficient salt as low NaCl intake exaggerates volume depletion and increases the secondary hyperaldosteronism resulting in intense hypokalemia [2, 3]. The patients is doing well, at least that is what we found on his last outpatient follow-up in June 2009. The prognosis of Gitelman syndrome is favorable because it is a chronic benign disease, and a lifelong nephrological, cardiovascular or endocrinological dispensarization is inevitable, thus mandating long-term evaluation. Overall, no complications occurred in treated patients [7, 19-21, 24, 25].
In summary, Gitelman syndrome is one of the rare causes of hypokalemia and it seems a challenge for physicians. In spite of being a congenital disorder, salt-losing tubulopathy may present in adulthood and should be borne in mind in the diagnosis of hypokalemia. Our patient is an example of Gitelman syndrome presenting with severe hypokalemia and periodic paralysis. Assessment of serum electrolytes including magnesium, evaluation of renal potassium and calcium excretion, acid-base analysis and presence of secondary hyperaldosteronism are essential in the approach to the patients with hypokalemic paralysis. If possible, diagnoses were performed at the genetic level. Dynamic studies with diuretic challenge test may be of diagnostic help when mutational study is not available. Suitable treatment protects patients from potentially dangerous complications.
Acknowledgments
We acknowledge the late Dr. H. Pudji Rahardjo, nephrologist, for his help with expert opinion on the renal clearance test. Most importantly, we would like to thank the patient.
Conflicts of Interest
The authors declare that they have no conflicts of interest related to the contents of this article.
Funding
This case report did not receive any specific grant from any funding agency in the public, commercial or non-profit sector. The authors did not receive any funding from any organization.
Author Contributions
FS and RMN both contributed equally to writing the manuscript, researching the discussion, reviewing and editing the draft manuscript. WHS was the senior author and was responsible for the supervision, obtaining patient’s consent, reviewing and editing the final manuscript herewith submitted for publication.
| References | ▴Top |
- Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221-235.
pubmed - Shaer AJ. Inherited primary renal tubular hypokalemic alkalosis: a review of Gitelman and Bartter syndromes. Am J Med Sci. 2001;322(6):316-332.
doi pubmed - Nakhoul F, Nakhoul N, Dorman E, Berger L, Skorecki K, Magen D. Gitelman's syndrome: a pathophysiological and clinical update. Endocrine. 2012;41(1):53-57.
doi pubmed - Unwin RJ, Capasso G. Bartter's and Gitelman's syndromes: their relationship to the actions of loop and thiazide diuretics. Curr Opin Pharmacol. 2006;6(2):208-213.
doi pubmed - Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. 1996;12(1):24-30.
doi pubmed - Balavoine AS, Bataille P, Vanhille P, Azar R, Noel C, Asseman P, Soudan B, et al. Phenotype-genotype correlation and follow-up in adult patients with hypokalaemia of renal origin suggesting Gitelman syndrome. Eur J Endocrinol. 2011;165(4):665-673.
doi pubmed - Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB. Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59(2):710-717.
doi pubmed - Bettinelli A, Bianchetti MG, Girardin E, Caringella A, Cecconi M, Appiani AC, Pavanello L, et al. Use of calcium excretion values to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. J Pediatr. 1992;120(1):38-43.
doi - Elisaf M, Siamopoulos KC. Fractional excretion of potassium in normal subjects and in patients with hypokalaemia. Postgrad Med J. 1995;71(834):211-212.
doi pubmed - Futrakul P, Yenrudi S, Futrakul N, Sensirivatana R, Kingwatanakul P, Jungthirapanich J, Cherdkiadtikul T, et al. Tubular function and tubulointerstitial disease. Am J Kidney Dis. 1999;33(5):886-891.
doi - Kageyama K, Terui K, Shoji M, Tsutaya S, Matsuda E, Sakihara S, Nigawara T, et al. Diagnosis of a case of Gitelman's syndrome based on renal clearance studies and gene analysis of a novel mutation of the thiazide-sensitive Na-Cl cotransporter. J Endocrinol Invest. 2005;28(9):822-826.
doi pubmed - Palmer BF. A physiologic-based approach to the evaluation of a patient with hypokalemia. Am J Kidney Dis. 2010;56(6):1184-1190.
doi pubmed - Greenlee M, Wingo CS, McDonough AA, Youn JH, Kone BC. Narrative review: evolving concepts in potassium homeostasis and hypokalemia. Ann Intern Med. 2009;150(9):619-625.
doi pubmed - Colussi G, Bettinelli A, Tedeschi S, De Ferrari ME, Syren ML, Borsa N, Mattiello C, et al. A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol. 2007;2(3):454-460.
doi pubmed - Konrad M, Weber S. Recent advances in molecular genetics of hereditary magnesium-losing disorders. J Am Soc Nephrol. 2003;14(1):249-260.
doi pubmed - Tajima T, Kobayashi Y, Abe S, Takahashi M, Konno M, Nakae J, Okuhara K, et al. Two novel mutations of thiazide-sensitive Na-Cl cotransporter (TSC) gene in two sporadic Japanese patients with Gitelman syndrome. Endocr J. 2002;49(1):91-96.
doi pubmed - Aoi N, Nakayama T, Tahira Y, Haketa A, Yabuki M, Sekiyama T, Nakane C, et al. Two novel genotypes of the thiazide-sensitive Na-Cl cotransporter (SLC12A3) gene in patients with Gitelman's syndrome. Endocrine. 2007;31(2):149-153.
doi pubmed - Lin SH, Shiang JC, Huang CC, Yang SS, Hsu YJ, Cheng CJ. Phenotype and genotype analysis in Chinese patients with Gitelman's syndrome. J Clin Endocrinol Metab. 2005;90(5):2500-2507.
doi pubmed - Tang NL, Hui J, To KF, Ng HK, Hjelm NM, Fok TF. Severe hypokalemic myopathy in Gitelman's syndrome. Muscle Nerve. 1999;22(4):545-547.
doi - Cheng NL, Kao MC, Hsu YD, Lin SH. Novel thiazide-sensitive Na-Cl cotransporter mutation in a Chinese patient with Gitelman's syndrome presenting as hypokalaemic paralysis. Nephrol Dial Transplant. 2003;18(5):1005-1008.
doi pubmed - Akinci B, Celik A, Saygili F, Yesil S. A case of Gitelman's syndrome presenting with extreme hypokalaemia and paralysis. Exp Clin Endocrinol Diabetes. 2009;117(2):69-71.
doi pubmed - Nishihara G, Higashi H, Matsuo S, Yasunaga C, Sakemi T, Nakamoto M. Acute renal failure due to hypokalemic rhabdomyolysis in Gitelman's syndrome. Clin Nephrol. 1998;50(5):330-332.
pubmed - Pela I, Materassi M, Seracini D, Lavoratti G, Bettinelli A. Hypokalemic rhabdomyolysis in a child with Bartter's syndrome. Pediatr Nephrol. 2005;20(8):1189-1191.
doi pubmed - Bettinelli A, Tosetto C, Colussi G, Tommasini G, Edefonti A, Bianchetti MG. Electrocardiogram with prolonged QT interval in Gitelman disease. Kidney Int. 2002;62(2):580-584.
doi pubmed - Scognamiglio R, Calo LA, Negut C, Coccato M, Mormino P, Pessina AC. Myocardial perfusion defects in Bartter and Gitelman syndromes. Eur J Clin Invest. 2008;38(12):888-895.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Nephrology and Urology is published by Elmer Press Inc.